 case report
case report AMOTL1 De Novo Mutation Associated with Orofacial Clefting, Ocular Colobomas, an Atrial Septal Defect and Digital Anomalies: A Case Report and Review of the Literature
Ashna Shome1*, Allysa Tuite2, Beth A. Pletcher3
1Medical Student, Rutgers New Jersey Medical School, Newark, New Jersey, USA
2Department of Pediatrics, Rutgers New Jersey Medical School, Newark, New Jersey, USA
3Department of Pediatrics, Rutgers New Jersey Medical School, Newark, New Jersey, USA
*Corresponding author: Ashna Shome, Medical Student, Rutgers New Jersey Medical School, Newark, New Jersey, USA
Received Date: 08 November 2022
Accepted Date: 16 November 2022
Published Date: 19 November 2022.
Citation: Shome A, Tuite A, Pletcher BA (2022) AMOTL1 De Novo Mutation Associated with Orofacial Clefting, Ocular Colobomas, an Atrial Septal Defect and Digital Anomalies: A Case Report And Review of the Literature. Arch Pediatr 7: 223. DOI: https://doi.org/10.29011/2575-825X.100223
Abstract
AMOTL1 is a membrane associated protein that has been described in 3 previous case reports involving craniofacial, cardiac, and musculoskeletal abnormalities. We present the fifth case of similar abnormalities associated with an AMOTL1 mutation. This patient is a two-year-old female with cleft lip and palate, left postaxial foot polydactyly, an atrial septal defect, unilateral iris and chorioretinal colobomas, facial dysmorphisms and speech delay. She was found to have a mutation in AMOTL1 (c.470G>A, p.(R157H). This report suggests a broad range of congenital defects associated with mutations in AMOTL1, and indicates the need to further characterize these mutations, with the goal of facilitating better screening and management of such patients.
Keywords: AMOTL1; Cleft lip and palate; Motilin; Multiple congenital anomalies
Introduction
AMOTL1, or angiomotilin-like 1, is a membrane associated protein in the motilin family associated with a variety of functions including cellular permeability, motility, and angiogenesis. Three previous case reports have described common features of craniofacial abnormalities and congenital cardiac abnormalities associated with an AMOTL1 mutation. Other features have included tethered spinal cord, imperforate anus, and congenital diaphragmatic hernia. All three cases have reported normal intelligence with speech delay likely related to oral clefts. These reports suggest that there is a wide range of phenotypic effects associated with these likely pathogenic mutations.
The first published case related to an AMOTL1 mutation described a father-son pair. The father was born with unilateral cleft lip and palate, with an atrial septal defect (ASD) repaired in childhood and an unknown mitral valve abnormality. The son was diagnosed prenatally with a ventricular septal defect (VSD) and cleft lip and palate. He was later also found to have tetralogy of Fallot and a double orifice mitral valve. A large cisterna manga was incidentally identified and remained unchanged since original imaging. Additionally, the patient had dysmorphism of the fingers, with a camptodactyly noted at rest. Both father and son were tall with dysplastic ears. The father was found to have a missense mutation (c.469C>T) in a highly conserved sequence, which was passed to the son [1].
The second case described a seven-month-old female born with hypotonia, cleft lip and palate, imperforate anus with rectovaginal fistula, pilonidal dimple and dysmorphic features including hypertelorism, micrognathia, large prominent ears with a folded earlobe, elongated fingers, and hallux varus of the right foot. Further evaluation showed choroid plexus cysts and a tethered cord. Development was age appropriate. The mutation was identified as a de novo missense variant (c.479C>T) in a highly conserved residue [2].
The most recent case describes a twenty-year-old male born with an ASD, congenital diaphragmatic hernia, cleft lip and palate, and facial dysmorphism including large ears, midface retrusion, saddle nose, small upper lip, short philtrum and deeply set eyes. He has speech delay with otherwise normal development. Sequencing showed a de novo heterozygous variant of uncertain significance (c.470 G>A) in AMOTL1 [3].
Here, we present a case of a two-year-old female with craniofacial, cardiac, and ophthalmologic abnormalities, with an associated AMOTL1 variant of uncertain significance identical to the twenty-year-old male in the previous report (c.470 G>A).
Case Presentation
The patient was delivered vaginally at 40 weeks gestation, and birth was complicated by maternal COVID-19 infection. She was admitted to the NICU for further management of the cleft lip and palate which had been observed on fetal anatomy scan. At birth the infant was observed to have left postaxial foot polydactyly. At birth, a heart murmur was appreciated, and the patient was found to have a small ASD. Karyotype and microarray showed normal results. The patient remained in the NICU for 4 days for feeding support.
On presentation, the patient was noted to have repaired cleft lip and palate, unilateral iris and chorioretinal colobomas, and facial dysmorphisms including hypertelorism, a broad and depressed nasal bridge, shallow orbits, OD exotropia and a short forehead [see figures 1 and 2]. Malocclusion of the maxillary and mandibular teeth was also observed. Developmental history at 19 months was significant for speech delay (only one-word utterances) and otherwise normal development with normal receptive language skills.
Whole exome sequencing performed at this time identified a de novo heterozygous variant of uncertain significance in AMOTL1 (c.470G>A, p.(R157H). The family was counselled on the possible significance of this mutation. Treatment course has included patching of unaffected eye, surgical repair of cleft lip and palate, removal of her postaxial extra toe, early intervention for speech delay, and continued monitoring for her ASD.
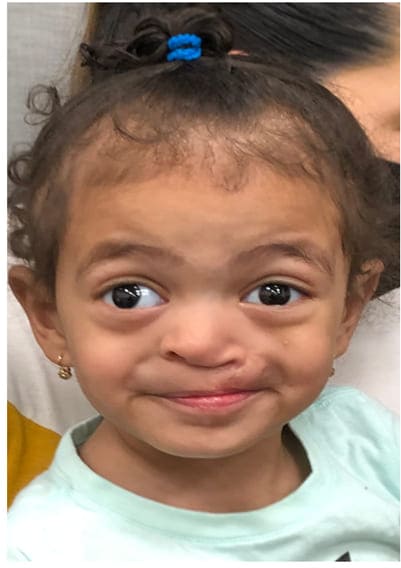
Figure 1: Facial features of case subject, including hypertelorism, broad and depressed nasal bridge, shallow orbits, short forehead, malocclusion of maxillary and mandibular teeth, repaired cleft lip and palate, OD exotropia. Reproduced with permission from her family.
Figure 2: Profile of case subject.
Discussion
The four cases described here of craniofacial and cardiac defects along with facial dysmorphisms suggest that an AMOTL1 mutation could be responsible for this multiple congenital anomaly phenotype (Table 1). All case subjects were overall developmentally appropriate with normal intelligence. Notably, all four cases have varied organ system involvement, suggesting a broad possible phenotypic range associated with mutations in a similar region of AMOTL1, which is known to be highly conserved (see Table 1). This suggests the very strong likelihood of a functional domain implicated in organ development being altered in all four cases.
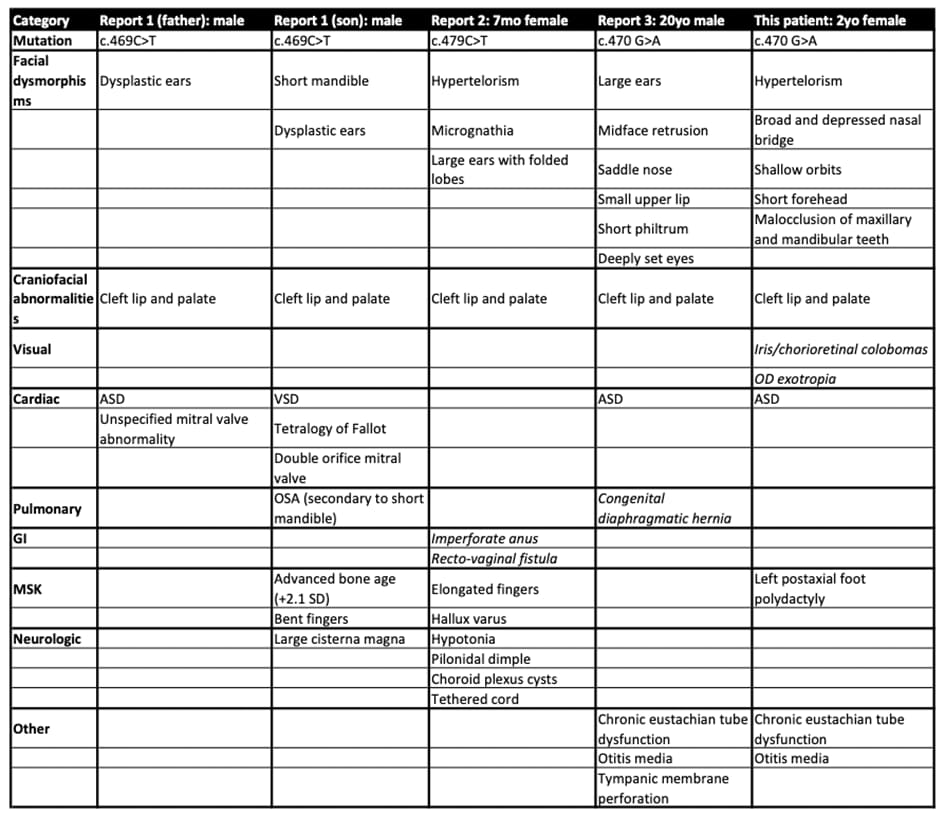
Table 1: Five case reports describe phenotypes with AMOTL1 mutation; Similarities between the cases include craniofacial dysmorphisms, cardiac involvement (ASD, VSD, Tetralogy of Fallot), and abnormal digits; Report #2 describes unique features of GI and neurologic involvement; Report #3 describes a unique congenital diaphragmatic hernia; Our patient has unique involvement of the eyes with iris and chorioretinal colobomas and OD exotropia.
All four cases described a de novo heterozygous mutations, with one case of transmission from father to son [1]. This suggests a possible autosomal dominant pathogenic mutation. Two identified cases have the same mutation (c.470G>A) in our report and one published by Lee&Gooch in 2019. The patient described here had unique ophthalmologic involvement, while the previously described case was significant for congenital diaphragmatic hernia. This supports our assertion that the mutation is pathogenic with a broad phenotype.
Our report adds to a growing body of knowledge that suggest a wide variety of congenital defects associated with autosomal dominant pathogenic mutations in AMOTL1. All five reported patients share congenital cardiac defects, orofacial clefting, as well as facial and limb dysmorphisms. However, the patient reported here has unique ophthalmologic involvement represented by an iris and chorioretinal colobomas as well as unilateral foot polydactyly. It is anticipated that further characterization of these mutations might to lead to better screening and management of such patients.
Acknowledgements
The authors would like to thank the patient and her family for their participation in this report.
References
- Liegel RP, Finnerty E, Blizzard L, DiStasio A, Hufnage RB, et al. (2019) Using human sequencing to guide craniofacial research. Genesis 57: e23259.
- Rips J, Mor-Shaked H, Erdin S, Yanovsky-Dagan S, Eventov-Friedman S, et al. (2021) De novo variant in AMOTL1 in infant with cleft lip and palate, imperforate anus and dysmorphic features. Am J Med Genet A 185: 190-195.
- Lee P, Gooch C (2022) eP168: A de novo variant in AMOTL1 gene in an adult with craniofacial abnormalities and previously unreported congenital diaphragmatic hernia. Genetics in Medicine 24: 1.